欢迎来到中国科学院海洋研究所科研物资管理系统
 DeepBio得分:5567.2
DeepBio得分:5567.2
 DeepBio得分
是基于文献引用次数,影响因子,文献新近度等因素计算的客观产品评级,得分越高表明该产品经过越可靠的实验验证,质量可信度越高
DeepBio得分
是基于文献引用次数,影响因子,文献新近度等因素计算的客观产品评级,得分越高表明该产品经过越可靠的实验验证,质量可信度越高
Hieff Clone® Plus One Step Cloning Kit是一款简便、快速、高效的DNA定向克隆产品,该试剂盒可以将PCR产物定向克隆至任何载体的任何位点,可高效克隆50 bp-10 kb片段。将载体线性化,并在插入片段正、反向PCR引物5’端引入15-25 bp的线性化载体末端同源序列,使得插入片段PCR产物5’和3’末端分别带有与线性化载体两末端对应的完全一致的序列。PCR产物和线性化载体在重组酶的作用下,仅需50℃反应20 min即可进行转化,完成定向克隆。克隆阳性率可达95%以上。
|
组分编号 |
组分名称 |
产品规格 |
|
|
10911ES20 (20 T) |
10911ES50(50 T) |
||
|
10911-A |
2×Hieff Clone® Enzyme Premix |
200 μL |
500 μL |
|
10911-B |
500 bp Control Insert (25 ng/μL) |
5 μL |
5 μL |
|
10911-C |
pUC 19 Control Vector, linearized (50 ng/μL) |
5 μL |
5 μL |
快速克隆;定向克隆;定点突变。
冰袋运输。-20℃保存,有效期1年。
1. 需自备的材料:
1)自备样品:自备好线性化载体和插入片段。
2)自备试剂(仅罗列部分):
① 超级感受态:转化效率>108 cfu/μg,如翌圣DH5α超级感受态细胞(Cat#11802);
② 高保真酶: 2×Hieff Canace® Gold PCR Master Mix(Cat#10149)或其他等效产品;
③ 菌落PCR mix:2×Hieff® Robust PCR Master Mix(With Dye)(Cat#10106)或其他等效产品;
④ 核酸染料:YeaRed Nucleic Acid Gel Stain (10,000 × in Water)(Cat#10202)或其他等效产品;
3)自备仪器耗材(仅罗列部分):PCR仪,水平电泳槽,切胶仪,EP管等;
2. 为了您的安全和健康,请穿实验服并佩戴一次性手套操作。
3. 本产品仅作科研用途!
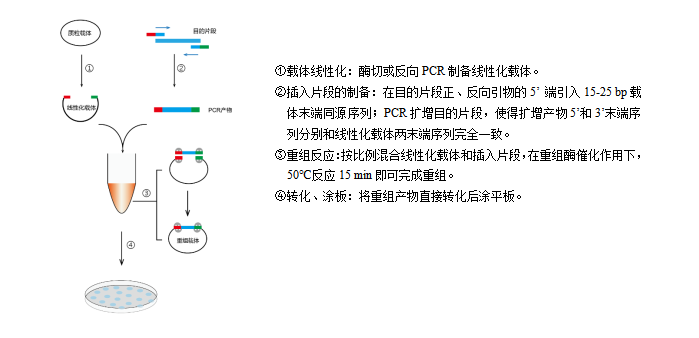
图1 Hieff Clone® 一步法快速克隆试剂盒实验流程图。
一、重组反应
1. 线性化载体与插入片段使用量计算
Hieff Clone® 重组反应体系最适载体使用量为0.03 pmol;最适载体与插入片段摩尔比为1:2-1:3,即最适插入片段使用量为0.06-0.09 pmol。这些摩尔数对应的DNA质量可由以下公式粗略计算获得:
最适载体使用量 = [0.02×载体碱基对数]ng (0.03 pmol)
最适插入片段使用量 = [0.04×插入片段碱基对数]ng (0.06 pmol) 或= [0.06×插入片段碱基对数]ng (0.09 pmol)
例如,将长度为2 kb的插入片段克隆至长度为5 kb的载体时,载体的最适使用量应为:0.02 × 5000 = 100 ng;插入片段最适使用量应为:0.04 × 2000 = 80 ng或0.06 × 2000=120 ng。
【注】:
2.重组反应体系(推荐冰上配制,各组分使用前需混匀)
|
组分 |
重组反应 |
阴性对照-1 |
阴性对照-2 |
阳性对照 |
|
ddH2O |
Up to 20 μL |
Up to 20 μL |
Up to 20 μL |
Up to 20 μL |
|
2×Hieff Clone® Enzyme Premix |
10 μL |
0 μL |
0 μL |
10 μL |
|
线性化载体 |
X μL |
X μL |
0 μL |
1 μL |
|
插入片段 |
Y μL |
0 μL |
Y μL |
1 μL |
3.重组反应条件
1)体系配制完成后,用移液器轻轻吸打混匀各组分,短暂离心将反应液收集至管底。
2)置于50℃反应20 min。当插入片段>5 kb时,可将孵育温度延长至25 min。
【注】:建议在PCR仪或水浴锅等温控精确的仪器上进行反应。
3)待反应完成后,建议将反应管置于冰上冷却5 min,以防温度过高降低感受态转化效率。
4)反应产物可直接进行转化,也可储存于-20℃,待需要时解冻转化。
二、 重组产物转化、涂板
1)在冰上解冻克隆感受态细胞(如:DH5α Chemically Competent Cell,Cat#11802)。
2)取10 μL冷却重组产物,加入到100 μL感受态细胞中,轻弹管壁数下混匀,在冰上放置25 min。
3)42℃热激45 sec,冰浴孵育1-2 min。
4)加入750 μL SOC或LB培养基,37℃孵育2 min充分复苏。37℃,200 rpm,摇菌60 min。
5)5000 rpm离心1 min,弃掉750 μL上清。用剩余培养基将菌体重悬,用无菌涂布棒在含有正确抗性的平板上轻轻涂匀。待菌液被吸收,将平板倒置,于37℃过夜培养。
最方便快捷的方法是菌落PCR。用无菌的枪头或牙签将单个菌落挑至20-50 μL LB培养基中混匀,直接取1 μL作为PCR模板。推荐至少用一条通用测序引物进行菌落PCR,这样可以避免PCR假阳性的产生。后续也可做酶切或测序鉴定。
1、最佳克隆位点选择?
答:在选择克隆位点时,应避免选择克隆位点上下游50 bp内有重复序列的区域。当克隆位点上下游25 bp区域内GC含量均在40%-60%范围内时,重组效率将达到最大。若高于70%或者低于30%,重组效率会受到较大影响。
2、无克隆长出或克隆数较少?
答:出现该情况,建议使用阳性对照,可排除试剂盒本身的影响,并进行进一步判定,主要有以下可能情况:
1)引物设计不合适:推荐【同源序列(15-25 bp)+完整的酶切位点+基因特异性扩增引物】,GC含量40% - 60%。
2)感受态细胞效率低:使用新鲜制备或妥善冻存的感受态细胞,确保其转化效率>107 cfu/μg。每次操作时可设置一组转化质粒的对照实验,以检测感受态细胞的转化效率。连接产物体积不应超过感受态细胞体积的1/10,否则会降低转化效率。
3)线性化载体和插入片段扩增产物的使用量不足/过量,或者比例不佳:尽量按照推荐的量和比例配制重组反应体系。
4)线性化载体和插入片段扩增产物不纯,抑制反应:线性化载体和插入片段扩增产物未纯化,直接使用时,使用总体积应不超过反应体系体积的1/5,如20 μL体系不超过4 μL。建议线性化载体、插入片段扩增产物进行凝胶回收纯化,纯化产物溶解在ddH2O中。
3、出现较多假阳性
答:主要有以下可能情况:
1)载体线性化不完全:即使是痕量未完全酶切的载体也会产生很高的转化背景。可通过阴性对照检测载体是否线性化完全,优化酶切体系,提高限制性内切酶使用量、延长酶切反应时间、胶回收纯化酶切产物等都可以有效减少环状质粒残留。
2)插入片段扩增产物混有非特异扩增产物:建议:①优化PCR体系,提高特异性;②胶回收PCR产物;③鉴定更多克隆。
3)反应体系中混入了相同抗性的质粒:PCR扩增模板(载体或插入片段)为环状质粒时,若扩增产物直接用于重组反应,残留环状质粒模板会产生较高的转化背景。建议使用预线性化质粒作为扩增模板、扩增产物进行DpnI消化或扩增产物进行胶回收纯化。
4、菌落PCR无条带
答:主要有以下可能情况:
1)引物不正确:推荐使用载体的通用引物或至少使用一条通用引物进行菌落PCR检测。
2)PCR体系或程序不合适:没有目的条带也没有空质粒条带,建议优化PCR体系、程序;或者提取质粒,以质粒做模板PCR验证;或者进行酶切验证。
3)重组失败:没有目的条带,只有空质粒的条带,说明重组不成功,载体线性化不完全,建议优化酶切体系。


 收 藏
收 藏